
小编:漂向北方丁爷
如果你曾送過歐洲與美國的註冊申請,你會發現有三種文件非常類似:設計歷史文件、510(k)提交以及技術文件。如果沒有送過,那這篇入門很適合你。趕快來看吧
設計歷史文檔
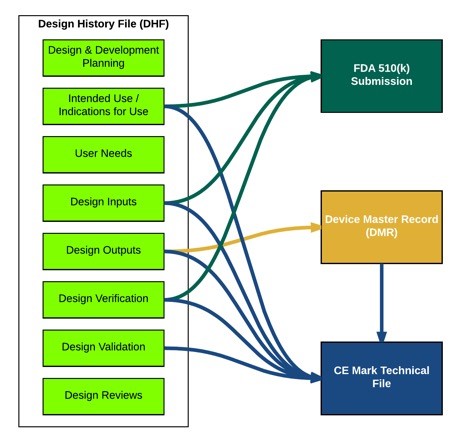
設計歷史文件是美國FDA 21 CFR Part 820.30中描述的術語,其中討論了設計控制以及如何將它們保存在設計歷史文件(DHF)中。 設計歷史文件就是設計和開發過程中收集的文件
根據FDA的規定,“每個製造商應為每種類型的設備建立和維護DHF。 DHF應包含或參考必要的記錄,以證明設計是按照批准的設計方案和本部分的要求制定的。
“ISO 13485:2003沒有直接提到DHF或類似的東西。 但是,更新後的ISO 13485:2016現在規定需要建立“設計和開發文件”
DHF現在有更多的重點,因為它被公認為包含設計控制文件的關鍵文件。 這很重要,因為您的設計控制是您的記錄,用來證明您的產品是安全的。設計計畫的製定是為了控制設計過程,然後生成一系列文檔來支持設計計畫 – 設計需求、設計結構、設計規格、驗證和確效。
多数厂商維護DHF通常是一個非常原始的過程,涉及老式三環活頁夾,文件櫃和紙張。 如果通過電子系統管理,DHF的創建變得更容易。
DHF構成了510(k)和技術文件的基礎,我們接下來分別探討510(k)和技術文件
510(k)提交
510(k)提交是醫療設備獲得FDA批准的共同途徑。大多數II類設備(和一些I類和III類;一些II類不要求510(k))需要510(k)。目的是向FDA證明您的產品基本上等同於市場上的產品。它包含20個部分,用於設備描述,使用指示和性能等不同的內容。您的設計控制將會進入許多這些部分。
許多公司沒有給自己一個既定的設計控制和DHF的良好基礎。在準備510(k)之前,這應該是你的第一步,因為它們提供了你的產品開發過程的文件證明,以證明你的設備是安全有效的。
一旦你有510(k)的許可,你可以在任何時候接受FDA檢查。您的設計控制和DHF將成為他們審查的主要議題,如果這些不符合標準,您最終可能會收到483條意見,並可能會發出警告信。
圖片來源:greenlight.guru
技術文件
技術文件比510(k)更接近於設計歷史文件,他是用於進入歐洲和世界其他地區的註冊文件。技術文件的目的是證明產品符合適用的要求和歐盟醫療器械(或IVD)的規定。
技術文件比510(k)的規定來得少,但是具有設定好的格式。設計驗證可以通過比較分析、模擬使用、動物研究和/或臨床調查來完成。技術文件需包含Verification和Validation,但是510(k)只需要Verification
技術文件與510(k)提交文件的一個重要區別是需要提供臨床評估報告。臨床評估報告涉及產品的臨床調查、風險,並概述任何可能需要的上市後臨床追蹤(PMCF)研究。這與設計驗證具有可比性並起到類似的作用。
最後一個差異就是技術文件是每個等級都需要提交的,但是510(k)卻大多在第二等級的註冊申請才會遇到。
記得:
• 儘早確定您的設備分類
• 建立良好的設計控制
• 數位化您的設計歷史文件,以便於更新
• 建立您的設備主記錄
文獻
1. Design History File vs. 510(k) vs. Technical File: What Do Medical Device Developers Need to Know?
Three (3) important technical file and 510k submission differences

DMR 的箭頭是不是該指向 510K 而不是CE啊?
讚讚